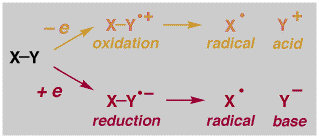
Ian R. Gould : Research Interests
Overview. In our lab we study various aspects of mechanistic organic and biochemistry, using the tools of photochemistry. We study both interesting chemical reactions, and use photophysical techniques to study processes that are difficult to investigate in other ways.
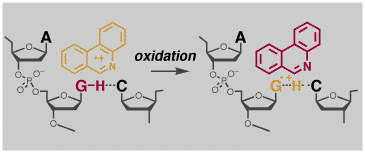
One main area of research involves studies of bond fragmentation reactions that are initiated by either giving (reduction) or taking away (oxidation) a single electron, as illustrated on the left below. Such processes generate radicals and Lewis acids and Lewis bases, which may also be Bronsted acids and bases, all potentially useful catalytic species. We are currently studying extremely fast fragmentation reactions with the goal of developing reactions that occur with no barrier at all! We are also using these reactions as test cases for fundamental reaction rate theories and contemporary computational methods. Another area relates to the oxidative processes in DNA responsible for mutation, disease and aging. Novel photochemical reactions developed in our lab can be used to irreversibly oxidize the purine bases in DNA (shown on the right, below). We are currently studying the chemistry and physical properties of these species in order to understand the primary events involved in oxidative damage.
|
|
Students involved in the work get the opportunity to learn a wide range of techniques and methods, including simple organic synthesis, steady-state and fast time-resolved laser emission and absorption spectroscopies to measure the kinetics of fast chemical reactions, computational chemistry methods and theory development, and simple biochemical analysis techniques.
Some More Details - Fragmentation Reactions. One fragmentation reaction we have studied in detail is that initiated by one-electron reduction of a series of N-methoxyheterocycles. The so-formed radicals undergo rapid N-O bond cleavage to yield a methoxy radical and the parent hetrocycle, with rate constant kfr. We measured values of kfr ranging over eight orders of magnitude, and some of the reactions occurred on a timescale of a few 100's of femtoseconds! One of these reactions is shown below.
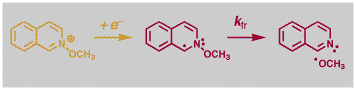
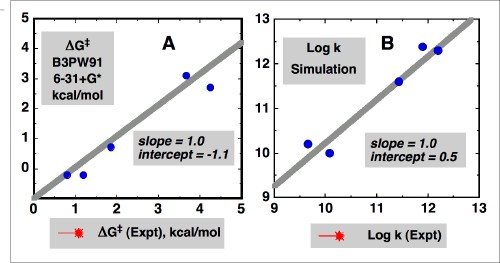
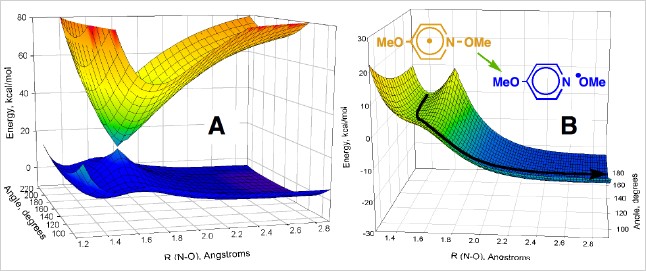
We were interested in exploring several broad areas of these and related reactions. First, we wanted to know if we could predict one of the most important pieces of information about any reaction, its reaction rate constant, e.g. kfr. We also wanted to know if we could design an electron transfer initiated bond-cleavage reaction that was barrierless, i.e. a reaction for which kfr was as high as possible. We were also interested in exploring the utility of contemporary electronic structure calculations. Finally we maintain an interest in relating experiment and theory that developed in previous work on electron transfer processes, and sought to apply this approach to "real" chemical reactions. A combined CASSCF/B3PW91 6-31+G* DFT study of the same molecules studied in solution (as opposed to models) revealed a remarkable correlation between experimental and DFT activation free energies (Figure). As shown on the left below, the computational and experimental activation free energies agree within ca. 1 kcal/mol. The reactions were further analyzed using a 3-D, two-state kinetic model including bond-stretching and bending coordinates, that gave reaction energy surfaces that were dominated by a conical intersection feature. Examples of these surfaces are shown on the right below for a p-styryl radical (A, both lower reactive and upper excited state surfaces shown) and the p-methoxypyridyl radical (B). A plot of the rate constants for the reactions obtained from these simulated energy surfaces versus experimental values exhibited a remarkably linear correlation (Figure). Our simple theoretical studies demonstrated how a barrierless reaction could be designed by an appropriate combination of reaction exothermicity and energy splitting of the diabatic states involved in the curve crossing. The p-methoxy radical (B) undergoes bond fragmentation with no energy barrier to the reaction at all. We are currently extending these studies to fragmentation reactions of charged radical ions in solution.
 |
 |
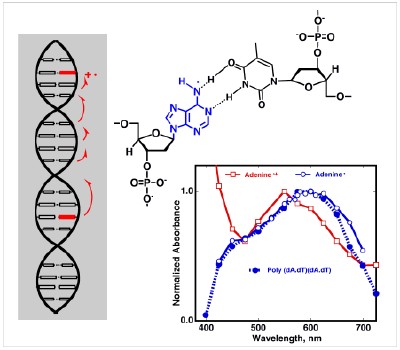
Some More Details - DNA Oxidation. Supported by the NSF, we have been looking at the primary processes in oxidative damage in DNA . Oxidation produces a radical cation of one of the purine bases (perhaps as a dimer or trimer), yet to date no such species has been directly observed in duplex DNA! We have developed the photochemistry of N-methoxyheterocyles as irreversible photooxidants. Excitation of N-methoxyphenanthridinium intercalated into poly (dA.dT)(dA.dT) irreversibly oxidizes the adenine. In this way the only transient species present after quenching the phenanthridine radical cation are the oxidized DNA products and a methoxy radical, which is transparent in the visible region. This allows detection of the weakly absorbing oxidized DNA products. A typical experiment is shown in the Figure below. The product of oxidation on the nanosecond timescale is the adenine radical, not the radical cation. Either the radical cation deprotonates rapidly, or alternatively, a proton-coupled initial electron transfer oxidation occurs. In either case it is clear that on the timescale of electron hopping or trapping by water, it is the purine radical, not the radical cation that should be considered. Further reactions of the oxidized purines may be "gated" by a reprotonation step, or again may involve proton-coupled electron transfer reactions. This is clearly a critical observation, since neither of these processes are currently considered in the long-distance electron transfer and DNA damage literature.
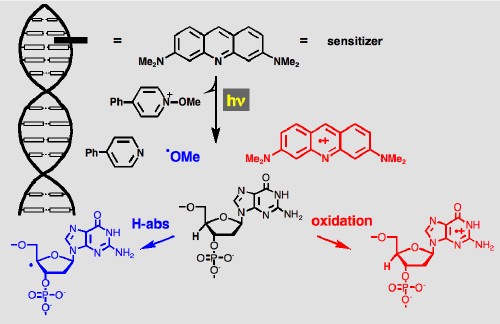
We are also using these reactions to study another fundamental problem in DNA. The two main mechanisms for single-strand cleavage in DNA involve hydrogen atom abstraction and oxidation, as indicated below. Electron transfer from a DNA intercalated excited state sensitizer to an N-methoxypyridinium forms the radical cation of the sensitizer (red), and a methoxy radical (blue), by fragmentation of the reduced pyridinium. The methoxy radical can initiate hydrogen atom abstraction, the radical cation can initiate oxidation of a DNA purine base. These two mechanisms can be selected based on the oxidation potential of the sensitizer, since use of a sensitizer with a lower oxidation potential makes the oxidation reaction endothermic. In this way, photochemical steady state and time-resolved methods have been used to quantify these different processes and their mechanisms. These studies are made possible because of the development of the photochemistry of N-methoxyheterocycles in our lab. Upon excitation these species are found to fragment from their excited singlet states to yield a methoxy radical and the radical cation of the heterocycle with high quantum yield (usually greater than 60%).
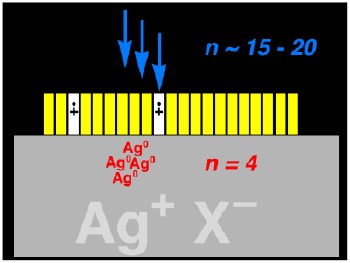
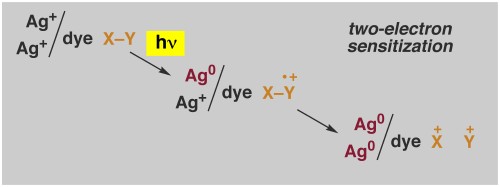
Some More Details - Chemistry of Photography. In collaboration with the Eastman Kodak Company, we have been working on one practical application of electron-transfer induced radical-ion fragmentation chemistry related to silver-halide photography. In conventional photography, light is absorbed by a monolayer of a sensitizing dye (yellow in the Figure). The excited dyes transfers an electron to a microcrystal of silver halide (usually a silver chloride/bromide). Remarkably, only 4 electrons need to be trapped in the silver halide crystal in the form of a silver atom cluster (red in the Figure) to capture an image that is revealed in the development process. Silver-halide photographic materials are extremely sensitive, and yet even more sensitive materials are always needed. Using radical cation fragmentation chemistry we have been able to almost double the sensitivity of photographic film
The approach, called Two-Electron Sensitization, is illustrated above. Light capture by a dye molecule as usual leads to the formation of an oxidized dye that in turn can oxidize an added donoir molecule, X-Y. The donor radical cation is designed to undergo controlled fragmentation to yield a cation and a radical X. The radical is also designed to be highly reducing, so that it injects a second electron into the silver halide, resulting in overall transfer of two electrons per absorbed photon. An overall reaction scheme is given below. The Two-Electron Sensitization technology has been used in several Kodak film products, mainly in the Motion Picture Area.
Ian R. Gould : Selected Publications
| Lorance, E. D., Hendrickson, K., Gould, I. R. "Density Functional Theory Accurately Predicts the Barriers for a Radical Reaction in Solution" J. Org. Chem., in press. |
| Lorance, E. D.; Kramer, W. H.; Gould, I. R. "Barrierless Electron Transfer Bond Fragmentation Reactions", J. Am. Chem. Soc., 2004, 126, 14071. |
| Gould, I. R.; Farid, S. "A Qualitative Curve-Crossing Model Describes the Kinetics of Oxidative Decarboxylation", J. Phys. Chem A. 2004, 108, 10949. |
| Morkin, T. L.; Turro, N. J.; Kleinman, M. H.; Brindle, C. S.; Griffiths, K.; Kramer, W. H.; Gould, I. R. "A Novel Solid-State Photooxidant", J. Am. Chem. Soc., 2003, 125, 14917. |
| Lorance, E. D.; Kramer, W. H.; Gould, I. R. "Kinetics of Reductive N-O Bond Fragmentation: The Role of a Conical Intersection" J. Am. Chem. Soc. 2002, 124, 15225. |
| Gould, I. R.; Lenhard, J. R.; Muenter, A. A.; Godleski, S. A.; Farid, S. "Two-Electron Sensitization: A New Concept for Silver Halide Photography", J. Am. Chem. Soc. 2000, 122, 11934. |
