![]()
![]()
Escapement Mechanism Hypothesis
For additional information concerning this hypothesis visit the simulations page.
We have found evidence that the F1Fo ATP synthase employs an escapement mechanism during ATP synthesis (8, and in the following paper). In this mechanism, the trans-membrane proton gradient provides constant torque to the g subunit (via the c-subunit ring). The interactions between the g and ab-subunit ring prevent this rotation until the empty catalytic site binds substrate. When the H-bonds and salt bridges at the catch loop are broken as the result of substrate binding, the torque on the g subunit is greater than the energy in the remaining H-bonds such that rotation of the g subunit induces the conformational changes in the catalytic sites necessary for ATP synthesis.
Zhou
et al.
(13)
demonstrated that substrate binding was a prerequisite of proton
gradient-driven rotation in E. coli F1Fo
by direct observation of rotation via the flag epitope. Mutations to residues in
the catch loop have been found in naturally occurring second site revertants of
the inhibitory gM23K mutant
(7)
. The gM23K
mutation was postulated to form an additional H-bond to the DELSEED region of
the b
subunit. We note that in every case, the reported revertants cause the loss or
weakening of a salt bridge or an H-bond either in the catch loop or near the g
subunit C-terminus. Second site mutations to restore ATP synthase activity to gQ269E
or gT273V
had similar effects and identified a third important location in the g
subunit N-terminus
(14)
. We note that several intersubunit H-bonds are present at the N-terminus
of the g
subunit. These results suggest that the sum of the energy in the intersubunit
H-bonds and salt bridges, regardless of their location, must not be above or
below a certain value for the enzyme to function.
If
the steps in ATP synthesis are the reverse of those during hydrolysis, the first
synthesis step involves a 30° rotation of the g
subunit that forms the tightly wound coiled coil. The available data suggests
that the intersubunit H-bonds and salt bridges appear to be stronger in this
conformation. Consequently, the proton-motive force may be capable of driving
the 30° rotation even though substrate has not bound to the empty catalytic
site. The energy of the proton gradient may be
sufficient to induce rotations greater than 30° that induce conformations of
the catalytic sites that are not conducive to product formation.
Interactions
between gR268,
gQ269
and the b
Subunit Catch-Loop of
E. Coli F1 ATPase are Important for Catalytic Activity †.
Matthew D. Greene and Wayne D.
Frasch*
†
This work was supported by National Institutes of Health Grant GM50202.
To download PDF of this paper from JBC click here
Summary
Removal of the ability to form a salt bridge or hydrogen bonds between the b subunit catch loop (bY297 - D305) and the g subunit of E coli F1Fo ATP synthase significantly altered the ability of the enzyme to hydrolyze ATP and the bacteria to grow via oxidative phosphorylation. Residues bT304, bD305, bD302, gQ269, and gR268 were found to be very important for ATP hydrolysis catalyzed by soluble F1-ATPase, and the latter four residues were also very important for oxidative phosphorylation. The greatest effects on catalytic activity were observed by the substitution of sidechains that contribute to the shortest and/or multiple H-bonds as well as the salt bridge. Residue bD305 would not tolerate substitution with V or S and had extremely low activity as bD305E, suggesting that this residue is particularly important for synthesis and hydrolysis activity. These results provide evidence that tight winding of the g subunit coiled coil is important to the rate-limiting step in ATP hydrolysis, and are consistent with an escapement mechanism for ATP synthesis in which abg intersubunit interactions provide a means to make substrate binding a prerequisite of proton gradient-driven g subunit rotation.
Introduction
The F1Fo
-ATP synthase1 uses a non-equilibrium transmembrane proton gradient
to catalyze the formation of ATP from ADP and inorganic phosphate. The enzyme
consists of two protein complexes, the membrane embedded Fo complex,
which couples proton translocation to the synthesis of ATP, and the membrane
extrinsic F1 complex, which contains the catalytic sites. The F1
portion consists of five subunits that occur with a stoichiometric ratio of (ab)3gde. The a and b subunits
are arranged alternately like the sections of an orange around a central g subunit stalk (see Fig. 1A). Catalytic activity
occurs at the ab interfaces, primarily within each
b subunit. The F1 portion can be isolated
from Fo and function as an ATPase
(1)
. The F1
functions as an ATP driven rotary motor
(2)
which moves through a
complete 360° rotation in three discrete 120° steps. The binding of Mg2+-ATP to a
catalytic site initiates a 90° rotation of the g
subunit to form an intermediate state. Following a 2ms pause, a 30° rotation concurrent with product release completes
the catalytic cycle
(3)
.
Differences in the conformation of the g
subunit have been observed between ground state F1 structures
(4,5)
and the (ADP×AlF4-)2F1
structure
by Menz et al
(6)
, a putative
post-transition state structure that contains Mg2+ADP and SO42-
at the low affinity catalytic site, and the transition state analog Mg2+-ADP-fluoroaluminate
bound at the other two catalytic sites. In this intermediate state structure the
position of the g subunit used to attach the probe for rotation studies is
rotated about 30° from its position in ground state structures. As a
consequence, the coiled coil of the g subunit is more tightly wound in the (ADP×AlF4-)2F1 structure
than in the ground state.
The major specific interaction between the helical coiled coil of the g subunit and the (ab)3 subcomplex occurs with gR2682
and gQ269
in all F1 structures. These residues form a ‘catch’ with a loop
of the b
subunit in the empty catalytic site conformation that encompasses residues
297-305 (Figure 1A). In the ground state structure, this catch results from a
salt bridge between gR268 and bD302 and from H-bonds between gQ269,
bD302
and bT304
(Figure 1B). In the (ADP×AlF4-)2
F1 structure, the catch loop of the catalytic site with bound
Mg2+-ADP and SO42- also interacts with gR268
and gQ269.
However, the interactions of these residues with the catch loop in (ADP×AlF4-)2
F1 differ from the ground state, such that gR268
and gQ269
are rotated about 15° as shown in Figure 1C. Among other differences, gR268
forms a second salt bridge with bD305 of the catch loop in (ADP×AlF4-)2F1.
The region of the g
subunit that interacts with the b subunit catch loop was one of three g
subunit locations where second site mutations suppressed the deleterious effects
of ATP synthase activity caused by the F1 gM23K mutant
(7)
. Based on these
observations, Al-Shawi et al
(7)
concluded that gM23K
decreases the coupling efficiency of F1Fo
due to an increase in the energy of interaction between b
and g
subunits. Catch loop residue bY297 is about 5.5 Å from the terminal phosphate in the
conformation of the catalytic site that binds Mg2+-AMPPNP
(4)
. Site directed mutants of
this residue in Chlamydomonas
chloroplast F1 decreased Mg2+-ATPase activity and changed
the EPR spectrum of vanadyl bound as VO2+-ATP to the low affinity
catalytic site
(8)
. These changes indicated
that this functional surrogate for the Mg2+ cofactor used bY297
as a metal ligand during the initial binding of metal-nucleotide to the empty
catalytic site. Based on these observations it was proposed that intersubunit
H-bonds between the g subunit and the (ab)3 ring prevent rotation driven by the proton
gradient until the empty catalytic site binds substrate. Deformation of the
catch loop-g subunit interactions induced by substrate binding would
provide an escapement mechanism that would maintain tight coupling between the
proton-motive force and ATP synthesis.
In this study we have mutated gR268 and gQ269 as well as residues in the catch loop of the b subunit in order to assess the importance of the intersubunit interactions at this location to catalytic activity. The results presented here suggest that these subunit interactions are very important to ATP hydrolysis and synthesis.
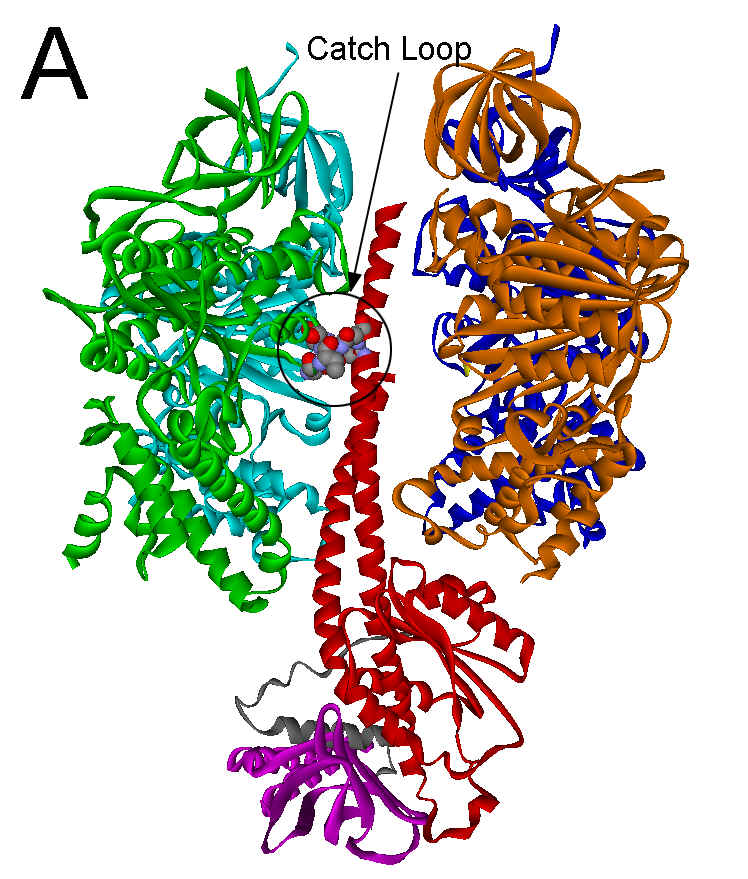
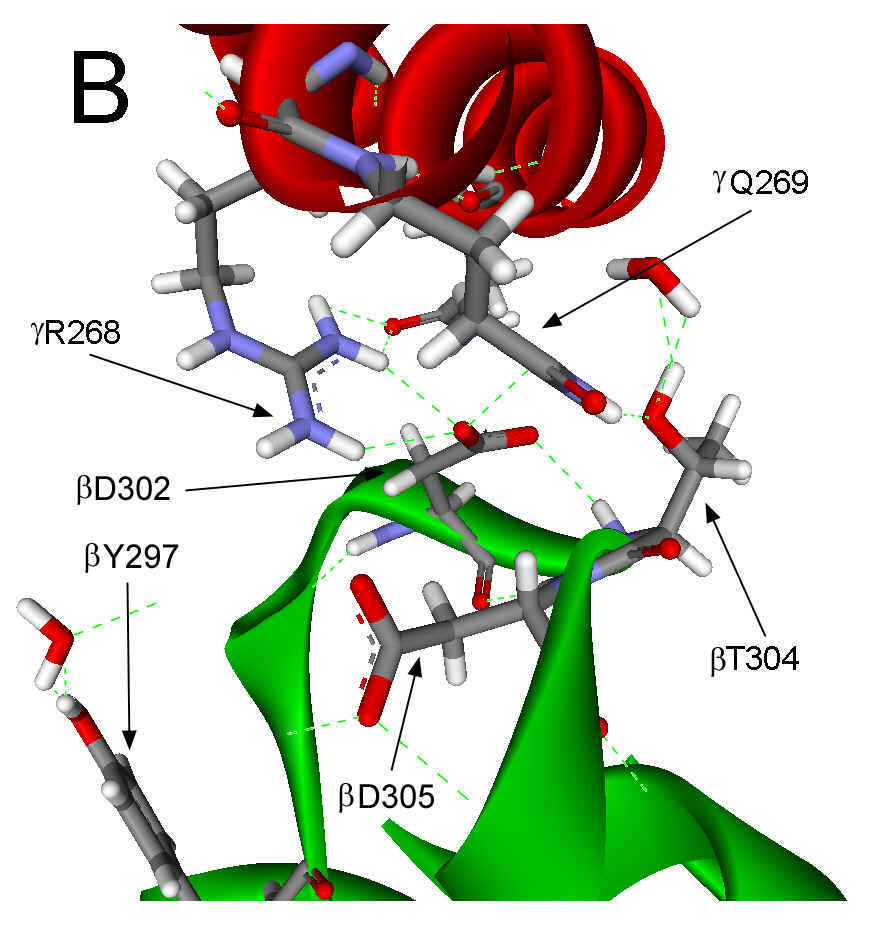
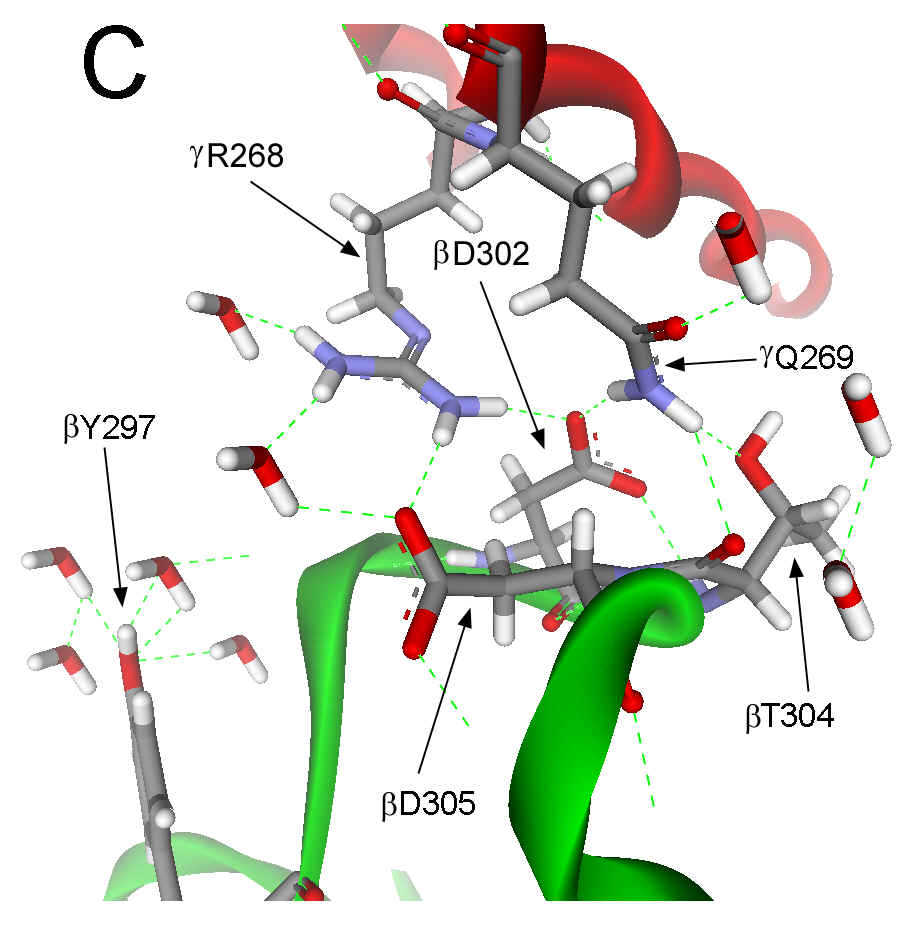
Figure 1. Interactions between the g subunit and the bE-catch loop of bovine F1-ATPase. (A) Location of the interactions between the catch loop of bE (green) and the g subunit (red) in the structure reproduced from Protein Data Bank file 1E79 using Web Lab Viewer from Molecular Simulations, Inc. Subunits aE and bTP are not shown for clarity. Details of the bE-catch loop-g subunit interactions in the ground state F1 structure (B) and the (ADP.AlF4-)2F1 structure (C) are reproduced from Protein Data Bank files 1E1Q and 1H8E, respectively, using Web Lab Viewer. Residues are labeled using E. coli numbering.
Experimental Procedures
Plasmid
p3U+ containing the entire E.
Coli unc operon and the AN887 strain of E.
Coli, which contains a Mu phage suppression of the endogenous unc operon,
were a generous gift from T. Duncan and R. Cross. Plasmid pLysS (Novagen) that
encodes T7 lysozyme was inserted to assist with breaking of the E.
Coli cells, and to provide chloramphenicol resistance. Transformation of the
AN887 strain by plasmid pLysS resulted in the strain ANLS. Construction of pXL1
was performed by insertion of a six histidine residue tag immediately after the
start codon of unc A, which encodes the F1 a subunit, using the
Stratagene Chameleon Double Stranded, Site-Directed Mutagenesis Kit. The
mutagenic primer was
5’TAAGGGGACTGGAGCATGCATCACCATCACCATCACCAGCTGAATTCCACCGAA-3’.
The italicized and underlined base insertions add a 6XHis tag, while the bold
base change introduces a PvuII site (CAA to CAG). The selection primer utilized was
5’-CTGTGACTGGTGACGCGTCAACCAAGTC-3’.
Here the italicized and underlined base changes convert the unique restriction
site ScaI to MluI (AGTACT to ACGCGT).
The desired mutations were first identified by screening with PvuII, and then
confirmed by DNA sequencing using ABI prism automatic sequencing, as were the
sequences of all other mutations. Strain AN887 was transformed via insertion of
pXL1 by electroporation
(9)
.
A further mutation, gS193C was made to facilitate attachment of rotation
probes for future studies. The mutagenic primer utilized was
5’-CTGCCGTTACCGGCATGCGATGATGATGATCTG-3’, and an XmnI restriction site was
deleted through the following mutagenic primer:
5’-CATCATTGGAAAACGCTCTTCGGGGCG-3’. The resulting cell line that contains the
6XHis tag, gS193C mutation, and XmnI deletion
is referred to as XL10.
The gR268L, gQ269L, bD302V,
bT304A, bD302T,
bD305V,
bD305S
and bD305E
mutants were prepared in plasmid pXL1 using the Stratagene Quick Change XL
Site-Directed Mutagenesis Kit with the oligonucleotide primers
5’-GGTATACAACAAAGCTCTTCAGGCCAGCATTACTCAG-3’,
and its complement 5’-CCTGAGTAATGCTGGCCTGAAGAGCTTTGTTGTATACC-3’
for creation of gR268L, 5’-GGTATACAACAAAGCTCGTCTGGCCAGCATTACTCAGG-3’,
and its complement 5’-CCTGAGTAATGCTGGCCAGACGAGCTTTGTTGTATACC-3’
for creation of gQ269L, 5’-GTATACGTACCTGCGGATGTCTTGACTGACCCG-3’
and its complement 5’-CGGGTCAGTCAAGACATCCGCAGGTACGTATAC-3’
for the creation of bD302V, 5’-CCTGCGGATGACTTGGCTGACCCGTCTCCG-3’
and its complement 5’-CGGAGACGGGTCAGCCAAGTCATCCGCAGG-3’
for creation of bT304A, 5’-GTATACGTACCTGCGGATACCTTGACTGACCCG-3’
and its complement 5’-CGGGTCAGTCAAGGTATCCGCAGGTACGTATAC-3’
for the creation of bD302T, 5’-GCGGATGACTTGACTGTTCCGTCTCCGGC-3’
and its complement 5’-GCCGGAGACGGAACAGTCAAGTCATCCGC-3’
for the creation of bD305V, 5’-GCGGATGACTTGACTAGCCCGTCTCCGGC-3’ and its complement 5’-GCCGGAGACGGGCTAGTCAAGTCATCCGC-3’
for the creation of bD305S, and 5’-GCGGATGACTTGACTGAACCGTCTCCGGC-3’ and its complement 5’-GCCGGAGACGGTTCAGTCAAGTCATCCGC-3’
for the creation of bD305E. The resulting plasmid was used to transform the ANLS
strain.
The F1-ATPase was purified from E. coli
using a modified form of the procedure described by Wise
(10)
. Cells were grown on LB
agar plates enriched with 10mM MgSO4 and 20mM glucose, containing 50 mg/mL ampicillin and 34 mg/mL chloramphenicol. Single colonies were picked and
transferred to 250mL flasks containing LB medium with the same supplements and
grown overnight at 37°C, and shaken at 250 rpm.
Overnight cultures were then transferred to 2L baffled flasks containing 1L of
minimal medium (60 mM K2HPO4, 40 mM NaH2PO4,
15 mM (NH4)2SO4) with 50 mg/mL ampicillin, 34 mg/mL
chloramphenicol and glucose to a final concentration of 30mM. Cells were grown
until late log phase at 37°C and shaken at 250 rpm.
The cultures were then harvested by centrifugation at 6000xg.
Prior to dissociation of F1 from Fo, membranes were
washed once with 50 mM TES (pH 8.0), 40mM eACA,
5%(v/v) glycerol, and 4.8 mM para-aminobenzamidine. Removal of F1
was accomplished via washing of membranes with 5 mM TES (pH 8.0), 40mM eACA,
5%(v/v) glycerol, and 1.0 mM EDTA. Dithiothreitol was excluded from all buffers
to avoid reducing the nickel column material. Membranes were then centrifuged at
100,000xg, and the supernatant containing F1 was concentrated down to
approximately 5 mL via pressure dialysis using an Amicon YM100 membrane. Nickel
affinity chromatography was utilized to purify the 6X His-tagged F1
under native conditions as described in the Qiagen Ni-NTA Superflow product
manual. Proteins removed from membranes in earlier steps were exchanged into
nickel column Binding Buffer (25mM Tris-HCl pH 8.0, 150mM NaCl, 10% glycerol,
1mM imidiazole, 1mM ATP). Buffer exchange was accomplished by concentrating the
crude F1 extract to less than 5% of the original volume by pressure
dialysis using an Amicon YM100 membrane followed by dilution in Binding Buffer.
Diluted F1 was stirred in the presence of Ni-NTA column material for
one hour, and packed into a column. Unwanted proteins were removed by flushing
the column in Wash Buffer (25mM Tris-HCl pH 8.0, 150mM NaCl, 10% glycerol, 10mM
imidiazole, 1mM ATP). The purified 6His-tagged F1 was then removed
with Elution Buffer (25mM Tris-HCl pH 8.0, 150mM NaCl, 10% glycerol, 100mM
imidiazole, 1mM ATP).
Succinate dependent growth measurements were determined by growing single
colonies picked from LB agar plates overnight at 37°C, and shaken at 250 rpm in
50mL cultures of minimal media described above with 50 mg/mL ampicillin, 34 mg/mL
chloramphenicol, 30mM succinate, and 600mg/L casein hydrolysate. Overnight
cultures were then inoculated into 1 L cultures of the same media, grown at 37°C and shaken at 250 rpm. Absorbance at 600nm was used
as a measure of cell density, and the doubling time of each culture was
determined in the log phase of growth.
The
rate of ATP hydrolysis was measured with an ATP regenerating coupled assay that
consisted of 50mM Tris-HCl pH 8.0, 10mM KCl, 2.5mM Phosphoenolpyruvate,
0.15mM-0.3mM NADH, 50mg/mL
pyruvate kinase, 50mg/mL
lactate dehydrogenase, 3nM F1 with 2mM Mg2+-ATP. The rate
was determined as the change in absorbance at 340nm using a Varian Cary 50 Bio
UV-Visible spectrophotometer equipped with a stirred cell Peltier temperature
control. The reaction was initiated by the addition of F1 to the
assay mixture. Reaction rates were calculated from data taken collected 8-10
minutes after initiation of the reaction to allow for dissociation of the e
subunit and to minimize inhibition by entrapped Mg2+-ADP
(11)
. Arrhenius analysis of data to
determine the entropic and enthalpic components of the free energy of activation
were performed using standard equations
(12)
:
EA = DH‡
+ RT
(Eq. 1)
DG‡
= DH‡
- TDS‡
(Eq. 2)
DG‡
= -RT ln (Nh/RT ×
kcat)
(Eq. 3)
where kcat is the turnover number, EA is the Arrhenius activation energy, N is Avogadro’s number, and DH‡, and DS‡ are the enthalpic and entropic components of the changes in Gibbs free energy of activation (DG‡). All Mg2+-ATPase assays were accomplished within 5 days of the date at which the F1-ATPase preparation was completed. After this period the preparations were found to lose activity as the result of an increase in the entropy of activation (data not shown).
Table
1. Comparison of the relative ability of mutant and XL10 strains to grow via
oxidative phosphorylation and of purified F1 ATPase to hydrolyze ATP.
|
Strain
Succinate-Dependent Doubling Timesa kcat
of F1-ATPase Activityd
(h)
(s-1) |
||
|
XL10 |
2.9 |
97 |
|
bD305V |
NGb |
--e |
|
bD305S |
NGb |
--e |
|
bD305E |
NGc |
4 |
|
bD302T |
4.3 |
3 |
|
bD302V |
3.9 |
0 |
|
bT304A |
2.7 |
30 |
|
gR268L |
4.7 |
12 |
|
gQ269L |
NGc |
1 |
a Conditions for growth on succinate via oxidative phosphorylation were the same as described in Experimental Procedures; b No growth was observed because F1Fo ATP synthase did not assemble; c No Growth observed after 13 h; d Conditions for ATP hydrolysis were the same as described in Experimental Procedures; e Not determined because F1-ATPase did not assemble.
Results
With the exception of bD305V and bD305S, the yield of F1 purified from the site-directed mutants to remove g subunit-catch loop interactions was approximately the same as that isolated from the XL10 strain. Intact F1-ATPase that contained the bD305V or the bD305S mutants was never successfully isolated and these mutations were presumed to interfere with assembly of the F1Fo ATP synthase. The F1-ATPase isolated from E. coli with the other mutations (gR268L, gQ269L, bD302T, bD302V, bT304A, or D305E) contained all five subunits as determined by SDS-polyacrylamide gel electrophoresis (data not shown). These results suggest that these latter mutations did not significantly affect the synthesis and assembly of the enzyme.
The relative ability of the mutant and wild type strains to grow via oxidative phosphorylation on minimal media in the presence of succinate was assessed by determining growth curves. The inability of bD305V and bD305S mutants to assemble intact F1 ATPase, coupled with their inability to grow on minimal medium with succinate was utilized as negative controls that the growth rate was dependent on the rate of ATP synthesis catalyzed by the F1Fo ATP synthase. The doubling times calculated from the growth curves are summarized in Table 1.
The F1Fo demonstrated very little tolerance for substitution of bD305. In addition to the fact that substitution of this carboxyl for a hydroxyl prevented assembly of F1, the most conservative mutation, bD305E, completely impaired the succinate-dependent growth rate. The strain that contained the gQ269L mutation was also unable to grow on succinate indicating that these residues are very important for ATP synthase activity. The ability of the strains that contained gR268L, bD302V, or bD302T mutants to grow on succinate was also significantly decreased. The doubling time of the strain containing gR268L was nearly twice that of XL10, while that of the strains that contained mutants of bD302 increased by about 1.5 fold. However, the rate of growth of the bT304A mutant on succinate was not affected. None of the mutations made to either the b or g subunits affected the ability of the bacteria to grow on minimal medium in presence of glucose. These results suggest that the poor growth on minimal media with succinate was the result of impairment of the ability of the F1Fo complex to synthesize ATP.
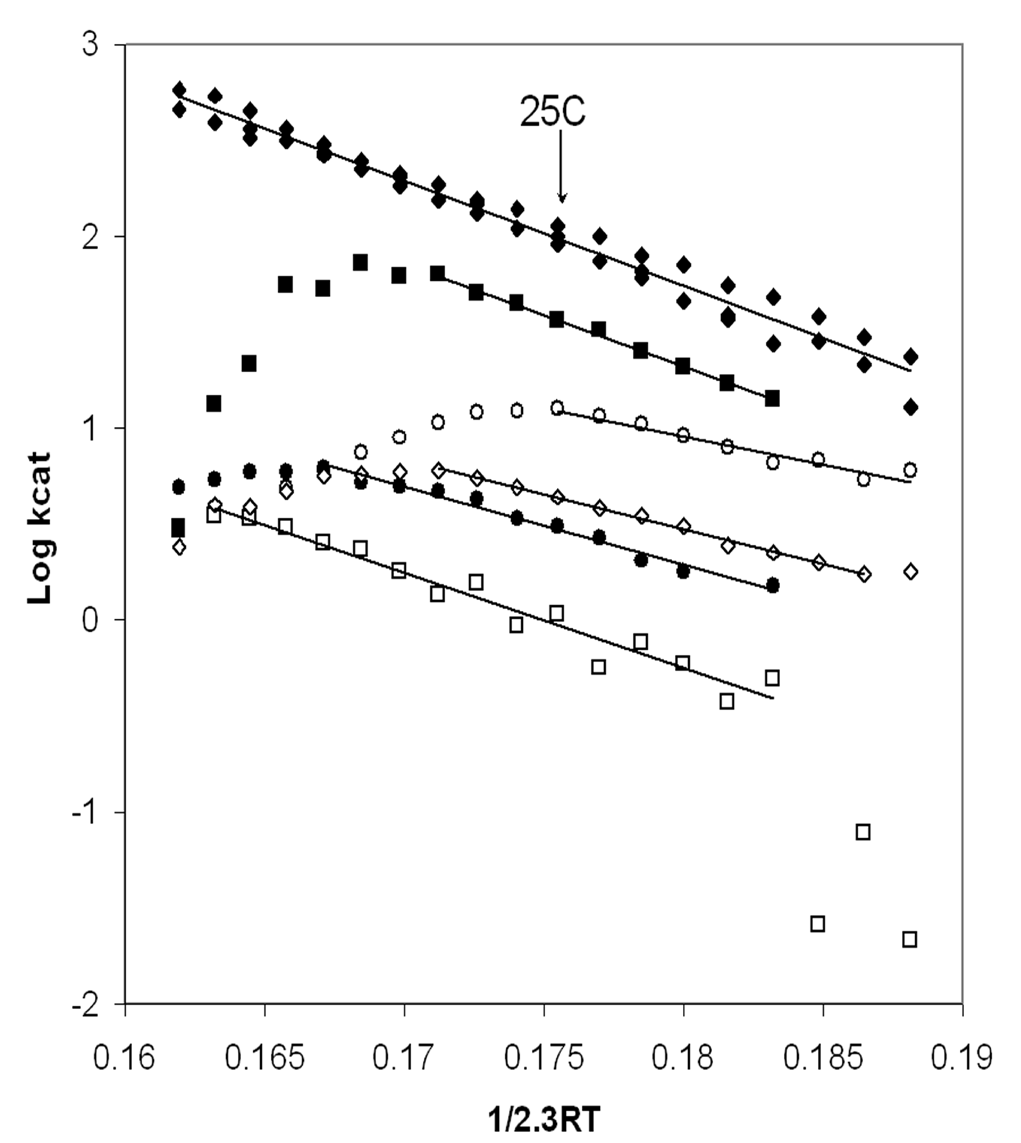
Figure 2. Arrhenius analysis of MgATPase activity catalyzed by soluble XL10-F1 (®), bD302T-F1 (l), bT304A-F1 (n), gR268L-F1 (m), bD305E-F1 (¯), and gQ269L-F1 (£). The concentration of F1 used was 3nM for XL10-F1, bD305E-F1, and bT304-F1 mutants, 9nM for the gR268L-F1, 40nM for gQ270L-F1, and 25nM for bD302T-F1. ATPase activities were assayed at 2mM Mg2+-ATP every 2.5°C from 5-50°C as described in Experimental Procedures. The lines plotted were generated by linear least squares regression of the data.
The mutations that interfered with g subunit-catch loop interactions also affected the ATPase activity of isolated F1 significantly. Figure 2 shows an Arrhenius plot the Mg2+-ATPase activity. With the exception of the gQ269L mutant, the mutations decreased the temperature stability of the enzyme. Although XL10-F1 was stable to 50°C, bT304A-F1 and bD305E-F1 were stable only to 32.5°C, while bD302T-F1 and gR268L-F1 reached their stability limits at 40°C and 27.5°C, respectively. Consequently, a direct comparison of the effects of the mutations on kcat was made at 25°C where all of the enzymes were stable as shown in Table 1.
The F1-ATPase that contained the bD302V mutation had no detectable ATP hydrolysis activity over the temperature range of 5- 50°C despite the fact that this mutant retained partial ability to grow on minimal media with succinate (Table 1). The more conservative bD302T mutation retained about 3 % of the XL10-F1 activity. The effect of this mutation was substantially greater than the effect of the mutation on succinate-dependent growth. The kcat values of bD305E-F1 and gQ269L-F1 were only 4% and 1 % of XL10-F1, respectively, which was comparable to their effect on the succinate dependent growth rate. It is noteworthy that the temperature stability of the gQ269L-F1 mutant was comparable to XL10-F1 despite the fact that both ATP hydrolysis and the growth rate on succinate were significantly lower than XL10-F1. The difference in ATPase activity and succinate dependent growth of the bT304A and gR268L mutations were 2.5-3 fold, with kcat values for ATP hydrolysis that were 45 % and 16 % of XL10-F1, respectively.
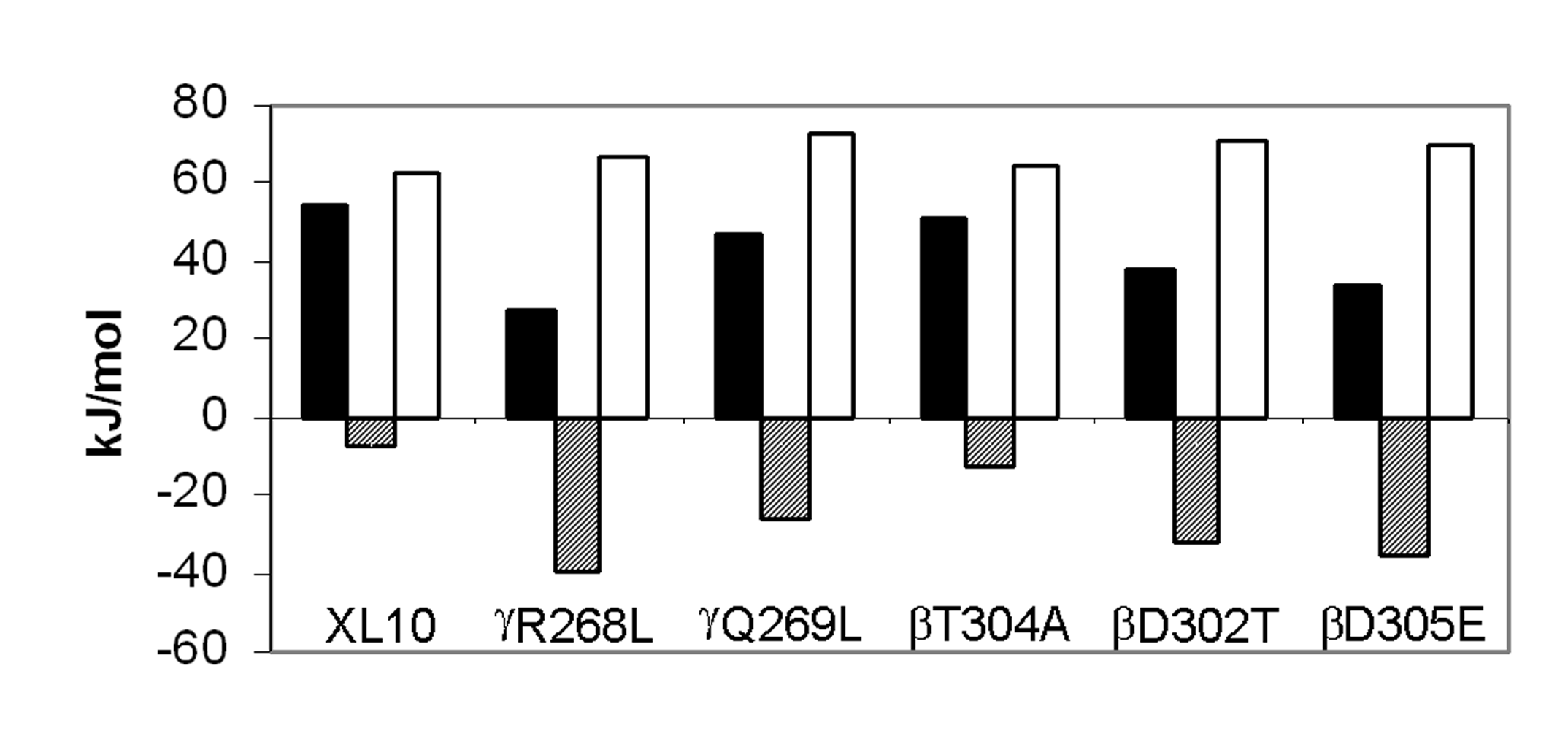
Figure
3. Comparison of thermodynamic parameters for steady state MgATP hydrolysis by F1
isolated from XL10 and from mutants that removed interactions between the g
subunit and the b subunit catch loop.
The parameters DH‡ (black
bars), TDS‡ (hatched bars),
and DG‡ (white bars) were
calculated at 25°C from the data
of Figure 2 as described in Experimental Procedures.
When the data are plotted as in Figure 2, the activation energy (EA) is determined directly from the slope. The values for enthalpy of activation, DH‡, of ATP hydrolysis are directly proportional to Ea as per Eq. 1, and were calculated from the data indicated by the solid lines in the Arrhenius plot as summarized at 25°C in Figure 3. For every mutant that retained Mg2+-ATPase activity, DH‡ was significantly lower than observed with XL10-F1. The gR268L mutation had the most dramatic effect on DH‡, with a value of 27.4 kJ/mol, which is approximately 50% of the observed XL10 value of 54.7 kJ/mol. Mutants bD305E and bD302T decreased DH‡ to 33.6 and 37.6 kJ/mol, or 61% and 68% of XL10 respectively. It is noteworthy that complete removal of the hydrogen bond and the salt bridge at this location via mutant bD302V rendered the enzyme unable to hydrolyze any ATP at all temperatures tested, and that the bD305S and bD305V mutants were unable to assemble F1Fo. Mutants gQ269L and bT304A also decreased the DH‡ of Mg2+-ATPase activity to about 46.9 kJ/mol and 51.1 kJ/mol, respectively.
Figure 3 also shows the entropy of activation, TDS‡, and free energy of activation, DG‡, derived from the data in Figure 2. The free energy of activation is inversely proportional to kcat as per Eq. 3. Since DG‡ is the result of the difference between DH‡ and TDS‡ (Eq. 2), the free energy barrier of the reaction will increase as the value of TDS‡ becomes more negative. Although all of the mutants lowered DH‡, lower rates of ATP hydrolysis were observed in every case because the entropic component of the energy barrier for the reaction increased (became more negative) to a greater extent than the decrease in DH‡. The value of TDS‡ for ATP hydrolysis of gR268L-F1 and bD305E-F1 was –39.4 kJ/mol, nearly a 6-fold difference from the XL10-F1 value of –7.5 kJ/mol. A 3.5 and 4.5-fold difference in TDS‡ from that of XL10-F1 was observed with bD302T-F1 (-32.7 kJ/mol) and gQ269L-F1 (-35.7 kJ/mol), respectively. Mutant bT304A, which retained its ability to grow on succinate and had the highest kcat for ATP hydrolysis, had the smallest changes in both DH‡ and TDS‡. The more negative values of TDS‡ observed in all of these mutants is consistent with an increase in disorder due to the weakening of, or loss of, either a hydrogen bond or salt bridge.
Discussion
The results presented here imply that hydrogen bonds and salt bridges between the b subunit catch loop and the g subunit are very important to the catalytic function of the enzyme. Residues bT304, bD305, bD302, gQ269, and gR268 were found to be very important for ATP hydrolysis catalyzed by soluble F1-ATPase, and the latter four residues were also very important for oxidative phosphorylation. At a resolution of 2.4 Å, typical of the available F1 crystal structures, it is difficult to determine if a hydrogen bond is present or assess its relative strength based on structural information alone. It is remarkable that single mutations to remove the possibility to make a single hydrogen bond or salt bridge outside the catalytic site can have such large effects on catalytic activity.
Several of the residues in the catch loop make a more important contribution to ATP hydrolysis than to ATP synthesis. The greatest differential effects were observed with mutants to bD302, although the gR268 and bT304 mutants also affect hydrolysis to a greater extent than synthesis. Even though mutations to these latter residues caused 2.5-3 fold differences in the decrease of hydrolysis and synthesis, the results indicate that gR268 was much more important to synthesis than bT304. These differential effects likely result from the fact that ATP hydrolysis was measured with isolated F1, while the competency of ATP synthesis was assessed by the growth rate of the E. coli strains on succinate. In the latter case, the fully assembled F1Fo ATP synthase may have a different rate-limiting step than isolated F1.
Relationship
between Catch Loop-g
subunit interactions and ATP hydrolysis. The rate-limiting step of the
ATPase reaction of isolated F1 occurs after a 90° rotation of the g
subunit induced by Mg2+-ATP binding
(3)
. The catalytic cycle is completed by a 30° rotation of the g
subunit concurrent with product release. In
the (ADP.AlF4-)2F1 crystal
structure, the portion of the g-subunit
used to attach the rotation probe is rotated about 30° from the ground state
structure
(6)
. This crystal structure contains Mg2+-ADP
and SO42- at the low affinity catalytic site, and the
transition state analog Mg2+-ADP-fluoroaluminate
at the other two catalytic sites.
Although
the foot region of the g-subunit,
the point of attachment for the rotation probe, was found to be rotated about 30°
in (ADP.AlF4-)2F1, the
C-terminus of this subunit is in nearly the same position as the other F1
structures. Consequently, the helical coiled coil of the g-subunit
is wound more tightly in (ADP.AlF4-)2F1
than the ground state structure. This implies that some of the torque on the g
subunit generated upon substrate binding may be used to wind the coiled coil
more tightly by inducing a 120° rotation of the C-terminus while the foot of
the g
subunit rotates 90°. Relaxation of
the coiled coil during the final 30° rotation could contribute to the energy
needed to complete the rate-limiting step of the reaction.
The
results presented here are consistent with a role for the intermolecular
interactions of the catch loop serving to promote the formation of the tightly
wound form of the coiled coil in a manner that provides energy for the final 30°
rotation during the rate-limiting step. Because product release is rate limiting
to F1-ATPase activity (3), the thermodynamic parameters measured here
provide information concerning this step. Despite the fact that all of the
mutants decreased DH‡,
the changes in entropy of activation more than compensated for the more
favorable values of DH‡,
thereby significantly lowering kcat. These changes in TDS‡
can be explained if the elimination of the salt bridges and/or H-Bonds between
the b
subunit-catch loop and g
subunit increased the number of allowable conformations of the F1
subunits during the rate-limiting step. Although some of these additional
conformations dramatically lower the activation energy barrier (and thus, DH‡)
from that of XL10-F1, many more of them are nonproductive. The
additional time needed for the enzyme to adopt a productive conformation in the
absence of the salt bridge or H-bond leads to the decrease in kcat.
It is noteworthy that bD305,
the residue that tolerated mutations least, forms a salt bride to the g
subunit in (ADP×AlF4-)2F1
that may represent the rate limiting conformation, but not in the ground state
structures.
Relationship between catch loop-g
subunit interactions and ATP synthesis. The results presented here also indicate that catch loop-g
subunit interactions are important to ATP synthesis. The succinate-dependent
growth rate of the gQ269L
and bD305E
strains was 1 % of wild type, suggesting that these residues were essential for
ATP synthase activity. The gR268L,
and both bD302
mutants also decrease the growth rate by 4 and 2 fold, respectively. These
results are consistent with the hypothesis that the residues at the catch loop
serve as an escapement mechanism during ATP synthesis (8).
In this mechanism, the trans-membrane proton gradient provides constant
torque to the g
subunit (via the c-subunit ring). The
interactions between the g
and ab-subunit
ring prevent this rotation until the empty catalytic site binds substrate. When
the H-bonds and salt bridges at the catch loop are broken as the result of
substrate binding, the torque on the g subunit is greater than the
energy in the remaining H-bonds such that rotation of the g
subunit induces the conformational changes in
the catalytic sites necessary for ATP synthesis. The results presented here
suggest that residues gQ269,
gR268,
bD305,
and bD302
contribute to the restraint of the rotation of the g-subunit
during ATP synthesis prior to the binding of substrate.
Zhou
et al.
(13)
demonstrated that substrate binding was a prerequisite of proton
gradient-driven rotation in E. coli F1Fo
by direct observation of rotation via the flag epitope. Mutations to residues in
the catch loop have been found in naturally occurring second site revertants of
the inhibitory gM23K mutant
(7)
. The gM23K
mutation was postulated to form an additional H-bond to the DELSEED region of
the b
subunit. We note that in every case, the reported revertants cause the loss or
weakening of a salt bridge or an H-bond either in the catch loop or near the g
subunit C-terminus. Second site mutations to restore ATP synthase activity to gQ269E
or gT273V
had similar effects and identified a third important location in the g
subunit N-terminus
(14)
. We note that several intersubunit H-bonds are present at the N-terminus
of the g
subunit. These results suggest that the sum of the energy in the intersubunit
H-bonds and salt bridges, regardless of their location, must not be above or
below a certain value for the enzyme to function.
If
the steps in ATP synthesis are the reverse of those during hydrolysis, the first
synthesis step involves a 30° rotation of the g
subunit that forms the tightly wound coiled coil. The available data suggests
that the intersubunit H-bonds and salt bridges appear to be stronger in this
conformation. Consequently, the proton-motive force may be capable of driving
the 30° rotation even though substrate has not bound to the empty catalytic
site. With the mutants described here, the energy of the proton gradient may be
sufficient to induce rotations greater than 30° that induce conformations of
the catalytic sites that are not conducive to product formation.
References
1. Frasch, W. D. (2000) Biochim Biophys Acta 1458, 310-325
2. Noji, H., Yasuda, R., Yoshida, M., and Kinosita, K., Jr. (1997) Nature 386, 299-302
3. Yasuda, R., Noji, H., Yoshida, M., Kinosita, K., Jr., and Itoh, H. (2001) Nature 410, 898-904
4. Abrahams, J. P., Leslie, A. G., Lutter, R., and Walker, J. E. (1994) Nature 370, 621-628
5. Braig, K., Menz, R. I., Montgomery, M. G., Leslie, A. G., and Walker, J. E. (2000) Structure Fold Des 8, 567-573
6. Menz, R. I., Walker, J. E., and Leslie, A. G. (2001) Cell 106, 331-341
7. Al-Shawi, M. K., Ketchum, C. J., and Nakamoto, R. K. (1997) J Biol Chem 272, 2300-2306
8. Chen, W., and Frasch, W. D. (2001) Biochemistry 40, 7729-7735
9. Sambrook, J., Fritsch, E.F., and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
10. Wise, J. G. (1990) J Biol Chem 265, 10403-10409
11. Kato, Y., Sasayama, T., Muneyuki, E., and Yoshida, M. (1995) Biochim Biophys Acta 1231, 275-281
12. Al-Shawi, M. K., and Senior, A. E. (1988) J Biol Chem 263, 19640-19648
13. Zhou, Y., Duncan, T. M., and Cross, R. L. (1997) Proc Natl Acad Sci U S A 94, 10583-10587
14. Nakamoto, R. K., al-Shawi, M. K., and Futai, M. (1995) J Biol Chem 270, 14042-14046
Footnotes
1Abbreviations: F1, The extrinsic membrane associated portion of the F1Fo ATP Synthase; Catch Loop, residues (bY297 - (bD305 of E Coli F1-ATPase; TES, 2-[(2-Hydroxy-1,1-bis(hydroxymethyl)ethyl)amino]ethanesulfonic acid; (ADP×AlF4-)2 F1, refers to PDB crystal structure H8E; ground state F1, refers to PDB crystal structure 1BMF or 1E1Q; eACA, e-aminocaproic acid, AMPPNP, adenosine 5’-(b,g-imino)triphosphate.
2E.
coli numbering of residues is used throughout this manuscript.
Acknowledgements
The authors thank Jennifer Sniegowski and David S. Lowry for assistance
with construction of mutants, Kathryn Boltz for development of methods, and
Laura Rodriguez for excellent technical support.